Bioinformática - Clustalw-MPI: Análise Filogenética utilizando computação paralela e distribuída
O software Clustalw-MPI é a ferramenta mais popular na implementação de múltiplo alinhamento de sequência de DNA. Sua principal diferença entre o software ClustalX dá-se a que ele possui uma melhor performance computacional devido realização do processamento em paralelo ou distribuído.
[ Hits: 21.381 ]
Por: José Cleydson Ferreira da Silva em 09/07/2010

Instalação do Clustalw-MPI
Sobre Clustalw-MPI
O software Clustalw-MPI é a ferramenta mais popular na implementação de múltiplo alinhamento de sequência de DNA. Sua principal diferença entre o software ClustalX dá-se a que ele possui uma melhor performance computacional devido realização do processamento em paralelo ou distribuído.A biblioteca utilizada na divisão do esforço computacional é chamada de MPI (Mensage Passing Interface). Ela proporciona redução no tempo de execução de determinadas processos.
O alinhamento pode ser obtido através de três estados:
- Alinhamento Pairwise
- Geração da arvore guia
- Alinhamento progressivo
O Clustalw-MPI é software semi-livre segundo a sua forma de licenciamento. Embora possua características de software livre como permissão de execução de programa, de estudo de seu funcionamento, de liberdade de redistribuição e de modificação, não há permissão de uso para fins comerciais. O autor do software chama-se Dr. Kuo-Bin Li <kbli@ym.edu.tw>, do instituto de bioinformática de Singapura.
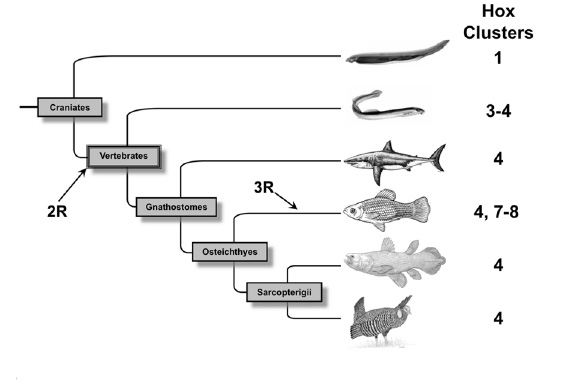
Performance Benchmark

Instalação do Clustalw-MPI
A instalação pode ser realizada de duas formas, através do gerenciador de pacotes da distribuição Debian ou derivados ou compilando através de código fonte. Segue abaixo as duas formas de instalação.Instalação via gerenciador de pacotes:
# apt-get install clustalw-mpi
Para se fazer a instalação por meio do código fonte é necessário verificar a configuração do arquivo Makefile. Os parâmetros de bem configurados podem proporcionar um bom funcionamento do software.
Parâmetros de configuração:
- CC = mpicc
- CFLAGS = -c -g
- or
- CFLAGS = -c -O3
Pode-se obter o código fonte do software no site do Instituto de Bioinformática de Singapura. Siga o processo de instalação por meio do código fonte no quadro abaixo.
Download via terminal:
# wget http://www.bii.a-star.edu.sg/docs/software/clustalw-mpi-0.13.tar.gz
Descompactar o arquivo:
# clustalw-mpi-0.13.tar.gz
Compilar o software:
# cd clustalw-mpi-0.13
# make
Um arquivo binário será gerado no diretório corrente um arquivo com o nome clustalw-mpi, entretanto pode-se movê-lo ou copiá-lo para o diretório /usr/bin para o que shell o identifique como um comando.
# mv clustalw-mpi /usr/bin/clustalw-mpi
ou
# cp clustalw-mpi /usr/bin/clustalw-mpi
Para se ter acesso ao executável que acabou de ser compilado, use o comando clustalw-mpi no terminal.
Usando clustalw-mpi
A usabilidade desse software dá-se por linhas de comando ou prompt de comando, a sintaxe da linha de comando determina usar os seguintes atributos.| mpirun | Executa tarefas paralelas em MPI. |
| -np | -npersocket, número de processos para cada nó. |
| 5 | Número de processadores. |
| clustalw-mp | Software que realiza o alinhamento de múltiplas sequências. |
| -Infile | Arquivos de entrada no formato FASTA. |
| -newtree | Arquivo de saída para gerar a árvore. |
Há 3 situações que caracterizam a usabilidade do Clustalw-MPI, que consiste em fazer alinhamento completo de múltiplas sequências, construir apenas árvore guia, múltiplo alinhamento de uma árvore existente e por fim a definição da variável de ambiente.
Alinhamento completo de múltiplas sequências
O alinhamento completo de múltiplas sequências usará um nó para mestre que por sua vez, irá gerenciar os processos enviados para os outros nós que se encontrem envolvidos no esforço de realização da determinada tarefa em paralelo.Usando por exemplo 10 processadores ou nós em rede, um será o gerenciador de envio e recebimento do resultado do processo e os outros 9 processadores ou nós serão utilizados para realizar os processamento da tarefa. A sintaxe de usabilidade desse contexto está representada.
# mpirun -cc 10 ./clustalw-mpi -infile=dele.input
# mpirun -np 10 ./clustalw-mpi -infile=CFTR.input
Árvore guia
Esse contexto refere-se apenas na criação de árvores, por meio de um arquivo de sequências. A sintaxe requer a indicação do arquivo de sequência e a definição de um novo arquivo onde será construída a arvore.# mpirun -np 5 ./clustalw-mpi -infile=dele.input -newtree=dele.mytree
# mpirun -np 5 ./clustalw-mpi -infile=CFTR.input -newtree=CFTR.mytree
Múltiplo alinhamento de uma árvore existente
Pode-se usar uma árvore já existente e fazer o alinhamento dela, para que isso ocorra é necessário usar o parâmetro -usetree.# mpirun -np 5 ./clustalw-mpi -infile=dele.input -usetree=dele.mytree
# mpirun -np 5 ./clustalw-mpi -infile=CFTR.input -usetree=CFTR.mytree
Variável de ambiente
A variável de ambiente a ser definida chama-se CLUSTALG_PARALLEL_PDIFF. A mesma é utilizada para executar alinhamento progressivo baseado em um pdiff paralelizado. Por padrão essa variável se encontra em estado não definida e o alinhamento progressivo será paralelizado conforme a estrutura da arvore de neighbor-joining.Habilitar essa variável requer entender a respeito de algumas funções, bem como parallelized pdiff(), prfalign(), e no entanto o manual do software não menciona como definir a variável CLUSTALG_PARALLEL_PDIFF. Deixá-la desabilitada não irá afetar a usabilidade e o desempenho do software.
2. Sobre o autor
Bioinformática - PhyML: alinhamento de sequências nucleotídicas em ambiente paralelo
Implementando servidor de aplicações PHP utilizando Zend Framework
Compiz - Janelas à 360 graus no Linux
Cairo-Dock - Seu desktop Linux com cara de MAC
Instalando um sistema tradutor de línguas no seu Linux
Sobre o GoblinX 1.1, mais programas e melhor rendimento
MySQL, Apache2, PHP5, phpMyAdmin e o driver de conexão com o NetBeans no OpenSUSE 11.2
Nagios 4 com Check_MK 1.2.5i3 no CentOS 6.5 x64
Produzindo um "reality show" com seu Linux
Cara, estou adorando essa serie de postagens sobre bioinformática. Sempre senti falta de alguma coisa no gênero em nossa lingua.
Muito bom!
Olá Diniz,
Realmente é uma boa oportunidade de apresentar essa série inedita de artigos em bioinformática. Espero postar outros artigos e apresentar outros softwares que fazem montagem de genona. Aproveite para indica-los a outros bioinformatas.
Obrigado, um abraço!
Olá Mauricio ('mecantao '),
O artigo realmente foi bem elaborado. Esse é um projeto que estou iniciando ainda em graduação, documentar grande parte dos softwares da área de bioinformática que são livres e de código aberto. Sempre dou preferência para aqueles que fazem processamento paralelo, o tempo gasto em analises de DNA são imensos e o paralelismo nos ajuda a diminuir esse tempo. Há outros artigos que escrevi e também uma comunidade aqui no viva o linux para tirar-mos dúvidas e postar-mos algumas novidades. Se estiver interessando entre na comunidade Open Source Bioinformatics.
Um abraço.
Patrocínio

Destaques
Artigos
Vale a pena ter mais de uma interface grafica no seu Linux?
Estrutura e Funcionamento de um Ebuild no Gentoo Linux
Instalação e Configuração do Void com Cinnamon
Dicas
Montagem pré automática de HD externo usb em NTFS não funciona no Debian Trixie - Solução
Guia de instalação do Gentoo Linux com Cinnamon (UEFI, LUKS2, Btrfs)
Tópicos
Top 10 do mês
-

Xerxes
1° lugar - 135.323 pts -

Fábio Berbert de Paula
2° lugar - 66.053 pts -

Buckminster
3° lugar - 42.149 pts -
Alberto Federman Neto.
4° lugar - 32.600 pts -

Alessandro de Oliveira Faria (A.K.A. CABELO)
5° lugar - 22.716 pts -

edps
6° lugar - 21.690 pts -

Daniel Lara Souza
7° lugar - 21.278 pts -

Mauricio Ferrari (LinuxProativo)
8° lugar - 20.387 pts -

Sidnei Serra
9° lugar - 19.806 pts -

Andre (pinduvoz)
10° lugar - 15.890 pts
Scripts
A maior comunidade GNU/Linux da América Latina! Artigos, dicas, tutoriais, fórum, scripts e muito mais. Ideal para quem busca auto-ajuda.
Site hospedado por: